
MITOFUSINA 2 – IDENTITY CARD
Il gene
La Mitofusina 2 è codificata dal gene MFN2 (conosciuto anche come HSG, MARF, CMT2A, CPRP1, CMT2A2), localizzato sul cromosoma 1 in posizione 1p36.22.
La regione genomica del gene MFN2 è lunga circa 33.000 paia di basi. La sequenza codificante del gene MFN2 è suddivisa in 19 esoni.

Sono state identificate due varianti trascrizionali, che codificano entrambe per la stessa proteina. La variante 1 (NM_014874) è lunga 4685 bp e rappresenta il trascritto più lungo. La variante 2 (NM_001127660) differisce dalla prima per una diversa regione non tradotta (UTR) a monte del sito di inizio della trascrizione ed è lunga 4540 bp.
Il gene MFN2 è espresso ubiquitariamente in tutti i tessuti.
La proteina
La Mitofusina 2 è una proteina transmembrana altamente conservata, ad attività GTPasica, localizzata sulla membrana mitocondriale esterna. È costituita da un grande dominio GTPasico e da due domini coiled-coil, tutti rivolti verso il lato citoplasmatico della membrana mitocondriale esterna.
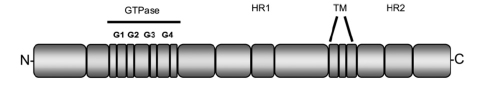
La funzione principale della Mitofusina 2 è la regolazione della fusione mitocondriale e l’interazione dei mitocondri con le membrane del reticolo endoplasmatico. Inoltre contribuisce al mantenimento e al funzionamento del network mitocondriale in cui questi organelli sono organizzati all’interno delle cellule. Infine, è coinvolta nella regolazione della proliferazione delle cellule della muscolatura liscia vascolare, e sembra giocare un ruolo nella patofisiologia dell’obesità.
IL RUOLO DELLA MITOFUSINA 2
Numerose ricerche hanno contribuito nel corso degli ultimi anni a chiarire il ruolo biologico della Mitofusina 2 nella fisiologia e nello sviluppo di malattie dell’uomo.
La sindrome di Charcot-Marie-Tooth (CMT) è fra le malattie ereditarie neurologiche più diffuse. Le mutazioni nel gene della mitofusina 2 (MFN2) causano il sottotipo assonale CMT2A, che è stato dimostrato, essere associato ad atrofia ottica, segni clinici del coinvolgimento del primo motoneurone, ed ictus ad esordio precoce. Le mutazioni MFN2 sono responsabili del 20-30% dei casi di CMT di tipo 2. Sono state riportate più di 50 mutazioni, trasmesse principalmente in modalità autosomica dominante, anche se sono state descritte famiglie con mutazioni omozigoti o eterozigoti composte. In famiglie di piccole dimensioni, mutazioni di MFN2 sono state associate con la gravità della neuropatia assonale, dell’atrofia ottica e del coinvolgimento del sistema nervoso centrale. I risultati delle biopsie di nervo evidenziano che i mitocondri sono strutturalmente anomali nei pazienti con mutazioni di MFN2.
Riportiamo di seguito alcuni contributi recenti sulle conseguenze per la salute umana di alcuni difetti molecolari identificati nel gene MFN2.
Il ruolo della Mitofusina 2 nel trasporto assonale dei mitocondri
Le alterazioni della dinamica mitocondriale (fissione, fusione e movimento) sono implicate in molte patologie neurodegenerative, dalle malattie genetiche rare come la sindrome di Charcot-Marie-Tooth, a condizioni più comuni come il morbo di Alzheimer. Tuttavia il rapporto tra le alterate dinamiche mitocondriali e la neurodegenerazione non è stato ancora completamente chiarito. Mutazioni nella proteina MFN2 identificate come patogenetiche nelle patologie umane sopprimono sia la fusione che il trasporto mitocondriale e producono le classiche caratteristiche della degenerazione segmentale assonale senza morte del corpo cellulare, che comprendono rigonfiamenti pieni di neurofilamenti, perdita dell’omeostasi del calcio e accumulo di specie reattive dell’ossigeno. Al contrario, la mancanza di Opa1 sopprime la fusione mitocondriale ma non il trasporto e non induce degenerazione assonale. La degenerazione assonale indotta dai mutanti MFN2 è dovuta ad un difetto nel posizionamento dei mitocondri all’interno degli assoni, piuttosto che alla perdita complessiva del loro movimento o ad una globale disfunzione mitocondriale. Abbiamo inoltre scoperto che aumentando l’espressione di MFN1 è possibile recuperare la degenerazione assonale causata dalla mutazione di MFN2, suggerendo così una possibile strategia terapeutica per la sindrome di Charcot-Marie-Tooth. Questi esperimenti dimostrano che la capacità dei mitocondri di percepire il fabbisogno energetico e di localizzarsi correttamente all’interno degli assoni è la chiave per il mantenimento dell’integrità assonale, e potrebbe essere un pathway attraverso il quale l’interruzione del trasporto assonale può contribuire alla neurodegenerazione.
[Mitofusin2 Mutations Disrupt Axonal Mitochondrial Positioning and Promote Axon Degeneration. J Neurosci. 2012 Mar 21;32(12):4145-55. Albert L. Misko, Yo Sasaki, Elizabeth Tuck, Jeffrey Milbrand, and Robert H. Baloh]
Le mutazioni
Mutazioni nel gene MFN2 associate alla condizione clinica atrofia ottica plus.
I geni MFN2 e OPA1 codificano per due proteine ad attività GTPasica “dynamin-like” coinvolte nella fusione della membrana mitocondriale. Questi due geni sono stati associati rispettivamente alla sindrome di Charcot-Marie-Tooth di tipo 2A e all’atrofia ottica autosomica dominante. In questa ricerca è stata studiata un’ampia famiglia con atrofia ottica esordita nella prima infanzia, associata a neuropatia assonale e miopatia mitocondriale in età adulta. Il quadro clinico si presenta come il fenotipo “plus” dell’atrofia ottica autosomica dominante legata a mutazioni in OPA1, ma è stato associato a una nuova mutazione missense di MFN2 (c.629A4T, p.D210V). Sono state riscontrate delezioni multiple del DNA mitocondriale (mtDNA) nel muscolo scheletrico e questo rende MFN2 un nuovo gene associato alla “sindrome da danno” del DNA mitocondriale. Contrariamente ai precedenti studi su pazienti con la sindrome di Charcot-Marie-Tooth di tipo 2A, i fibroblasti che portano la mutazione mostrano difetti della catena respiratoria, frammentazione del network mitocondriale e una significativa riduzione della proteina MFN2. Inoltre, per la prima volta la compromissione della fusione mitocondriale è stat correlata con un difetto nel sistema di riparazione dei danni al mtDNA indotti dallo stress. È probabile che il difetto nella riparazione dei danni al DNA mitocondriale sia dovuto ad una variabilità del contenuto di proteine all’interno della popolazione mitocondriale e può essere almeno parzialmente responsabile dell’instabilità del mtDNA.
[The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain. 2012 Jan;135(Pt 1):23-34. Cecile Rouzier, Sylvie Bannwarth, Annabelle Chaussenot, Arnaud Chevrollier, Annie Verschueren, Nathalie Bonello-Palot, Konstantina Fragaki, Aline Cano, Jean Pouget, Jean-Francois Pellissier, Vincent Procaccio, Brigitte Chabrol and Veronique Paquis-Flucklinger].
http://www.ncbi.nlm.nih.gov/pubmed/22189565
MITOL regola le interazioni tra reticolo endoplasmico e mitocondri, modulando l’attività della proteina Mitofusina 2
I mitocondri sono degli organelli cellulari estremamente dinamici continuamente soggetti a processi di fissione e di fusione. La proteina MITOL, localizzata a livello della membrana mitocondriale esterna, regolare le dinamiche mitocondriali interagendo con la proteina MFN2. La proteina MFN2 è abbondante a livello delle giunzioni tra reticolo endoplasmatico e mitocondri, definite “mitochondria-associated ER membrane (MAM)”, che sono essenziali per molte funzioni cellulari. In questo lavoro, gli autori hanno dimostrato il ruolo della proteina MITOL nel regolare l’interazione del reticolo endoplasmico e dei mitocondri, modulando l’attività della proteina MFN2 attraverso un processo di ubiquitinazione del residuo K192. La mancata espressione della proteina MITOL, infatti, altera la localizzazione della proteina MFN2 e la corretta funzionalità del complesso MAM. E’ quindi possibile che MITOL sia coinvolto nella patogenesi di malattie neurodegenerative, come la CMT2A, attraverso la regolazione della proteina MFN2. Futuri studi riguardo al ruolo della proteina MFN2, del dominio MAM e soprattutto della loro regolazione mediata da MITOL potrebbero contribuire ad incrementare la conoscenza dei meccanismi della CMT2A.
[Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, Inatome R, Yanagi S. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. 2013 Jul 11;51(1):20-34]
http://www.ncbi.nlm.nih.gov/pubmed/23727017
Mutazioni a livello della Mitofusina 2 alterano le funzioni mitocondriali attraverso deplezioni del DNA mitocondriale
La Charcot-Marie-Tooth di tipo 2a (CMT2A) è associata a mutazioni in eterozigosi a carico della proteina Mfn2, localizzata a livello della membrane mitocondriale e direttamente coinvolta nei processi di fusione mitocondriale. È ancora oggetto di discussione quale siano le conseguenze di queste mutazioni in termini di fosforilazione ossidativa. In questo lavoro, vengono studiati gli effetti funzionali delle mutazioni della Mfn2 in cellule muscolari e nei fibroblasti ottenuti da 4 pazienti CMT2A. Le disfunzioni osservate a livello della catena respiratoria mitocondriale potrebbero essere spiegate da un decremento del numero di copie del DNA mitocondriale (mtDNA). Infatti solo il paziente senza alterazioni a livello della catena respiratoria presenta anche un numero normale di copie del DNA mitocondriale. In particolare, gli autori hanno osservato alti livelli di delezioni del DNA mitocondriale nei pazienti CMT2A che sono più marcati nei pazienti senza deplezione del mtDNA. Analisi delle delezioni del mtDNA hanno rilevato la perdita di parti del dna mitocondriale richieste per la replicazione. Questa osservazione è in accordo con la mancanza di espansione clonale per la maggioranza delle delezioni del mtDNA osservate. Per quanto riguarda invece la riduzione del numero di copie, le delezioni inverosimilmente contribuiscono ad anomalie respiratorie. Dall’analisi di questi dati sembrerebbe che mutazioni a carico della proteina Mfn2 alterino la fosforilazione ossidativa a livello mitocondriale, influenzando la replicazione del mtDNA.
[Vielhaber S, Debska-Vielhaber G, Peeva V, Schoeler S, Kudin AP, Minin I, Schreiber S, Dengler R, Kollewe K, Zuschratter W, Kornblum C, Zsurka G, Kunz WS. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2013 Feb;125(2):245-56]