
MITOFUSIN 2 – IDENTITY CARD
THE GENE
Mitofusin 2 is encoded by MFN2 gene, also known as HSG, MARF, CMT2A, CPRP1, CMT2A2. By radiation hybrid analysis and by examining a human-rodent hybrid panel, Nagase et al. (1996) mapped the MFN2 gene to chromosome 1. Zuchner et al. (2004) stated that the MFN2 gene maps to chromosome 1p36.2
By sequencing clones obtained from a size-fractionated immature myeloid cell line cDNA library, Nagase et al. (1996) cloned this gene, which they designated KIAA0214. The deduced 757-amino acid protein contains an ATP/GTP-binding site motif. Northern blot analysis detected expression of MFN2 in all tissues and cell lines examined, with highest expression in heart and skeletal muscle.
When stably expressed in COS-7 cells, MFN2 colocalized with mitochondrial markers. An internal region, including the predicted bipartite transmembrane domain, was sufficient to target MFN2 to mitochondria. Casasnovas et al. (2010) noted that the MFN2 gene contains 19 exons.

Two transcriptional variants of MFN2 gene are known, differing for their untranslated region upstream the transcription start site.
THE PROTEIN
Mitochondria are not isolated organelles but rather form highly dynamic structures that continuously fuse and fragment. Together with Mfn1 and OPA1, Mfn2 plays a major role in the mitochondrial fusion process in mammalian cells.
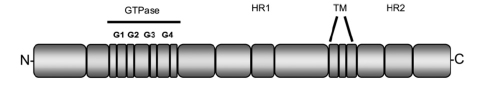
Mitofusin 2 (Mfn2) is one of the two mitofusin proteins also required for mitochondrial fusion. Mfn1 and Mfn2 are conserved integral outer mitochondrial membrane proteins, each consisting of a large GTPase domain and two heptad re- peats, or putative coil-coiled domains, all of which face the cytoplasm (Koshiba et al., 2004; Meeusen et al., 2004; Song et al., 2009).
THE ROLE OF MITOFUSIN 2
Charcot-Marie-Tooth (CMT) disease is amongst all the more commonly inherited neurological disorders (Skre et al.1974).
It comprises a group of disorders that affect peripheral motor and sensory nerves classically divided into demyelinating (CMT1) or axonal type (CMT2) according to nerve conduction velocity (Dyck PJ, Lambert EH, 1968, Reilly MM et al., 2011). Mutations in the mitofusin 2 gene (MFN2) result in the axonal subtype CMT2A. CMT 2A usually presents with progressive weakness and sensory loss. It has been reported also by our group that in some patients CMT2 may disclose a wider clinical spectrum including optic atrophy, clinical signs of first MN impairment, and CNS involvement (Del Bo R et al., 2008, Feely SM et al., 2011). MFN-2 is a nuclear encoded, dynamin-like protein, linked to the rearrangement of the outer mitochondrial membrane and tethering the endoplasmic reticulum to mitochondria. Several experimental data have shown that MFN-2 is a mediator of mitochondrial fusion, an evolutionarily conserved process responsible for the surveillance of mitochondrial homeostasis (Papanicolaou et al., 2011). Altered mitochondrial transport has been suggested to be a pathological mechanism of axonal CMT neuropathy, even if the exact causative processes are largely unknown (Misko et al., 2010).
Recent studies investigated the role of mutant Mitofusin 2 in human physiology and pathology.
Le mutazioni
Alterations in mitochondrial dynamics (fission, fusion, and movement) are implicated in many neurodegenerative diseases, from rare genetic disorders such as Charcot-Marie-Tooth disease, to common conditions including Alzheimer’s disease. However, the relationship between altered mitochondrial dynamics and neurodegeneration is incompletely understood. Here we show that disease associated MFN2 proteins suppressed both mitochondrial fusion and transport, and produced classic features of segmental axonal degeneration without cell body death, including neurofilament filled swellings, loss of calcium homeostasis, and accumulation of reactive oxygen species. By contrast, depletion of Opa1 suppressed mitochondrial fusion while sparing transport, and did not induce axonal degeneration. Axon degeneration induced by mutant MFN2 proteins correlated with the disruption of the proper mitochondrial positioning within axons, rather than loss of overall mitochondrial movement, or global mitochondrial dysfunction. We also found that augmenting expression of MFN1 rescued the axonal degeneration caused by MFN2 mutants, suggesting a possible therapeutic strategy for Charcot-Marie-Tooth disease. These experiments provide evidence that the ability of mitochondria to sense energy requirements and localize properly within axons is key to maintaining axonal integrity, and may be a common pathway by which disruptions in axonal transport contribute to neurodegeneration.
[Mitofusin2 Mutations Disrupt Axonal Mitochondrial Positioning and Promote Axon Degeneration. J Neurosci. 2012 Mar 21;32(12):4145-55. Albert L. Misko, Yo Sasaki, Elizabeth Tuck, Jeffrey Milbrand, and Robert H. Baloh]
http://www.ncbi.nlm.nih.gov/pubmed/22442078
Mutations in MFN2 gene resulting in a novel clinical presentation: autosomal dominant optic atrophy plus phenotype
MFN2 and OPA1 genes encode two dynamin-like GTPase proteins involved in the fusion of the mitochondrial membrane. They have been associated with Charcot-Marie-Tooth disease type 2A and autosomal dominant optic atrophy, respectively. We report a large family with optic atrophy beginning in early childhood, associated with axonal neuropathy and mitochondrial myopathy in adult life. The clinical presentation looks like the autosomal dominant optic atrophy ‘plus’ phenotype linked to OPA1 mutations but is associated with a novel MFN2 missense mutation (c.629A>T, p.D210V). Multiple mitochondrial DNA deletions were found in skeletal muscle and this observation makes MFN2 a novel gene associated with ‘mitochondrial DNA breakage’ syndrome. Contrary to previous studies in patients with Charcot-Marie-Tooth disease type 2A, fibroblasts carrying the MFN2 mutation present with a respiratory chain deficiency, a fragmentation of the mitochondrial network and a significant reduction of MFN2 protein expression. Furthermore, we show for the first time that impaired mitochondrial fusion is responsible for a deficiency to repair stress-induced mitochondrial DNA damage. It is likely that defect in mitochondrial DNA repair is due to variability in repair protein content across the mitochondrial population and is at least partially responsible for mitochondrial DNA instability.
[The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain. 2012 Jan;135(Pt 1):23-34. Cecile Rouzier, Sylvie Bannwarth, Annabelle Chaussenot, Arnaud Chevrollier, Annie Verschueren, Nathalie Bonello-Palot, Konstantina Fragaki, Aline Cano, Jean Pouget, Jean-Francois Pellissier, Vincent Procaccio, Brigitte Chabrol and Veronique Paquis-Flucklinger]
http://www.ncbi.nlm.nih.gov/pubmed/22189565
MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2
Mitochondria are cellular organelles constantly subject to highly dynamic processes, such as fission and fusion. The protein MITOL, localized to the outer mitochondrial membrane, regulates mitochondrial dynamics, interacting with the protein Mfn2. Mfn2 is enriched at the junction between the endoplasmic reticulum (ER) and mitochondria, which is known as the mitochondria-associated ER membrane (MAM), which is essential for many cellular functions. In this study, the authors have demonstrated the role of the protein MITOL about the interaction of the endoplasmic reticulum and mitochondria, by modulating the Mfn2 activity via K192 ubiquitination. Indeed, MITOL knockdown modifies Mfn2 localization, MAM function and their their regulation by MITOL. MITOL could be involved in the pathogenesis of neurodegenerative diseases, such as CMT2A, through Mfn2 regulation. Future studies about Mfn2 role, MAM domain and especially about their regulation mediated by MITOL could contribute greatly to our understanding of the mechanism of CMT2a.
Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, Inatome R, Yanagi S. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. 2013 Jul 11;51(1):20-34]
http://www.ncbi.nlm.nih.gov/pubmed/23727017
Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion
Charcot-Marie-Tooth neuropathy type 2A (CMT2A) is associated with heterozygous mutations in the mitochondrial protein mitofusin 2 (Mfn2) that is intimately involved with the outer mitochondrial membrane fusion machinery. The precise consequences of these mutations on oxidative phosphorylation are still a matter of dispute. Here, we investigate the functional effects of MFN2 mutations in skeletal muscle and cultured fibroblasts of four CMT2A patients. The observed dysfunction of the mitochondrial respiratory chain can be explained by a twofold decrease in mitochondrial DNA (mtDNA) copy numbers. The only patient without detectable alterations of respiratory chain in skeletal muscle also had a normal mtDNA copy number. We detected higher levels of mtDNA deletions in CMT2A patients, which were more pronounced in the patient without mtDNA depletion. Detailed analysis of mtDNA deletion breakpoints showed that many deleted molecules were lacking essential parts of mtDNA required for replication. This is in line with the lack of clonal expansion for the majority of observed mtDNA deletions. In contrast to the copy number reduction, deletions are unlikely to contribute to the detected respiratory impairment. Taken together, our findings corroborate the hypothesis that MFN2 mutations alter mitochondrial oxidative phosphorylation by affecting mtDNA replication.
[Vielhaber S, Debska-Vielhaber G, Peeva V, Schoeler S, Kudin AP, Minin I, Schreiber S, Dengler R, Kollewe K, Zuschratter W, Kornblum C, Zsurka G, Kunz WS. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2013 Feb;125(2):245-56]