
CLINICAL ASPECTS OF CMT2A
CMT 2A2: MITOFUSIN 2 GENE MUTATION-RELATED NEUROPATHY
(MFN 2, chromosome 1p36.22)
Epidemiology
CMT 2A2 affects around 19-23% of patients suffering from CMT2 type axonal neuropathy (inherited axonal neuropathy transmitted as autosomal dominant characteristic), thus representing the most frequent subtype of CMT2 type diseases. A study in 2006 published by the scientific journal Brain reported the percentage of affected patients as up to 33%.
Penetrance of the disease is variable, thus meaning that within the same families multiple occurrences can present with different clinical statuses in terms of characteristics and severity.
Inheritance
The Mitofusin 2 gene mutation responsible for axonal neuropathy is mainly transmitted as an autosomal dominant characteristic, i.e. the mutation carrier is a parent who will have a 50% chance of passing on the disease to the child.
In some rare cases the genetic transmission is homozygous recessive, i.e. both parents present with a MF2 gene mutation. In such cases the child will have a 25% chance of being healthy, a 25% chance of receiving both mutations and a 50% chance of receiving one mutation. Parents and children carrying one mutation can be either asymptomatic or presenting with mild neuropathy.
There are also compound trans-heterozygous mutations associated with a pattern of autosomal recessive transmission. These often determine a more severe, early-onset phenotype of the disease (Nicholson GA et al, 2008; Vallat JM et al., 2008; JM Polke et al., 2011).
CMT 2A2 can also result from de novo mutations, with no precedent family history.
Onset
CMT 2A2 onset is variable, ranging from the first decade (2-3 years) up to the fifth decade. Generally, these extreme early onset cases are the most severe.
Clinical history
The clinical history of CMT 2A2 is similar to the one of other inherited CMT2-related neuropathies (such as CMT2A1, CMT2E and CMT2F) which involve other genes.
In general, the primary symptom is a slow-progression motor and sensory inherited neuropathy. This begins in lower limbs and then expands to upper limbs, causing difficulties in deambulation, weakness in movement and difficulties in hand use.
The clinical history can vary, ranging from early onset cases with severe weakness to asymptomatic cases (up to 25% of all MTF2 gene mutations carriers present with subclinical signs only).
The range of signs and symptoms includes:
> Polyneuropathy
- distal weakness in hands and feet (affecting mostly legs rather than arms). This results in difficulties in deambulation and can cause complete deambulation impairment:
- hypotonia and reduction or absence of osteotendinous reflexes; frequent fatigue
- abnormalities in sensitivity
> Optic atrophy (in most severe cases)
> Pain, cramping
> Tremors
> Hearing loss
> Macrocephaly
> Psychomotor impairment
> Pes cavus, scoliosis
The presence of some of those characteristics can be related to specific Mitofusin 2 mutations. To date, scientists have discovered more than 80 mutations responsible for the disease.
Overall, based on the clinical status we can define:
- An early onset form (within the first decade), which is characterised by a severe phenotype and rapid course including loss of deambulation autonomy (generally within the second decade) leading to an early need for deambulation supports. With regard to neurophysiology, this subtype is associated with severe reduction in the amplitude of the compound muscle action potential (CMAP). The reduction is so significant that in some cases CMAP cannot be detected. There is evidence for this form being associated to the following mutations: L92P, R94W, R94Q,T105M, R364W (Zucher et al., 2004; Chung et al., Brain 2006)
- A late onset form (after the first decade), which is characterised by a less severe phenotype and a less aggressive course. Neurophysiologic studies show normal or mildly impaired CMAP. There is evidence for this form being associated with the following MFN2 gene mutations: M367T, H165R.
Clinical variants of standard CMT2A2 form
- HMSN V/CMT V + spasticity in lower limbs (variant including myelopathy)
Autosomal dominant transmission with onset starting from the second decade. Clinical history is consistent with standard CMT2 (weakness in lower limbs) with pyramidal signs (lively osteotendinous reflexes, plantar cutaneous response in extension, with or without spasticity). The clinical status is completed by abnormalities in sensitivity and pain. The disease progression is slow and most patients present with mild disability. - HMSN VI/CMTVI + optic atrophy
Autosomal dominant transmission or sporadic case with onset in childhood years. This variant is often associated with most severe phenotypes and de novo mutations. Clinical history present with a severe motor neuropathy (which is consistent with CMT) and optic atrophy with variable onset (from 5 to 50 years). The optic atrophy is progressive in children and slower in adults, and includes subacute loss of visual acuity with centrocecal scotomata, colour vision impairment and pale disk (which can be detected by an examination of the fundus oculi). In some cases the pathology can improve over time with different levels of regression and partial recovery of the visual acuity (as observed in some cases of Leber’s inherited optic neuropathy). - CMT + severe encephalopathy
Neuroimaging examinations identify signal hyperintensity in the cerebral peduncles, periventricular white matter and centrum semiovale. These can be found both in late onset forms (KW Chung et al., Brain 2006) and in early onset forms, and sometimes they are associated with secondary macrocephaly (R104W mutation) and microstructural alterations (astrogliosis), increase in neuroaxonal density and demyelination identified by cerebral SPECT scan (Brockmann et al., Journal of Neurology 2008).There are also “multiple sclerosis-like” clinical traits which are associated with MF2 gene mutations. Those are characterised by presence of progressive optic atrophy (mono and bilateral) and NMR-detectable alterations of brain signalling, with just mild clinical signs of axonal neuropathy which often occur at a later stage (mutazione Leu146Phe, Christopher J Klein et al., Archives of Neurology 2011).
- Mitochondrial myopathy with multiple deletions in the mitochondrial DNA caused by MFN2 mutations. The onset is in adult age and it is preceded by optic atrophy with onset in childhood and consequent development of axonal neuropathy (D210V mutation, Cecile Rouzier et al., Brain 2012).
- Recessive axonal neuropathy (AR-CMT2)
This form, with childhood onset, is more severe and has a progressive course leading to deambulation loss. It can present with optic atrophy and scoliosis. Electrophysiological examination shows absence of amplitude and speed reduction in CMAP and SAP with normal NCV.
Other variants
Fast progressive severe form with childhood onset (2-3 years), characterised by diffused weakness and sensitivity loss up to deambulation loss.
With regard to genetic profile, patients affected by this variant can present with homozygous mutations (same mutation on both alleles) or compound heterozygous mutations (two different mutations on the two alleles). Parents are asymptomatic or present mildly symptomatic (mild neuropathy).
Variant characterised by cognitive impairment with cerebral abnormalities in association with axonal neuropathy.
Diagnosis
Neurophysiologic examination
ENG/EMG is performed to study motor and sensory nerve conduction velocities. Those are mildly reduced in axonal neuropathies such as CMT2, with significant reductions in CMAP and SAP. Those results suggest axonal neuropathy.
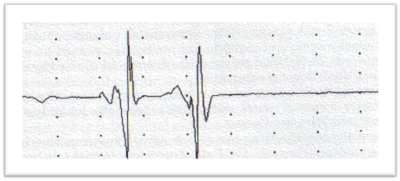
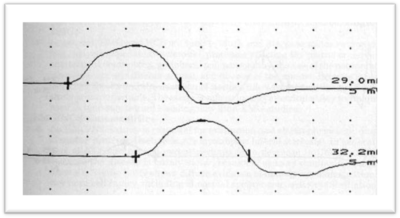
Genetic examination
When clinical and neurophysiologic data show evidence compatible with a CMT2A2 form, genetic examination is performed with Mitofusin gene sequencing.
Encephalic NMR can be useful in case of CNS-related symptoms; ophthalmological examination with examination of the fundus oculi and of the field of vision can be useful in case of visual acuity reduction; also, in cases of unclear diagnosis a sural nerve biopsy can be performed to gather additional morphostructural and immunohistochemical information on the peripheral nervous system.
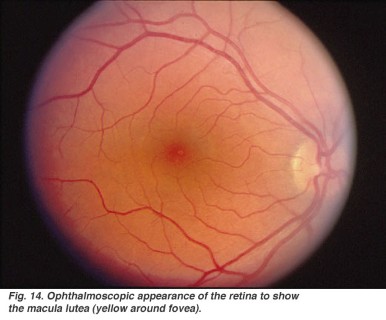
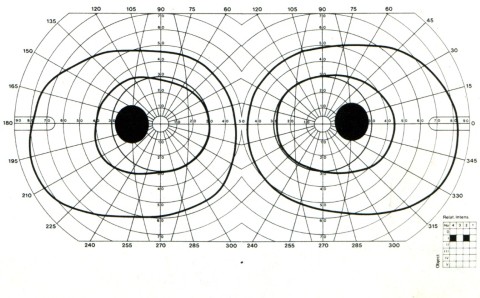
Treatment
To date there is no cure to CMT 2A2, and the only treatment that proved to be capable toimproving functional performances (e.g. in prehension or dehambulation) in patients affected by CMT is rehabilitation therapy.
Essential tools for rehabilitation treatment in CMT patients are orthopaedic supports such as shoes (either normal shoes specifically modified by an orthopaedist or custom-made shoes), insoles and supports to stabilise the ankle and avoid the foot dangling whilst walking.
Physiotherapy is important to prevent joint deformities and to improve functional performances after the adoption of appropriate shoes and orthopaedic supports.
Orthopaedic surgery may be useful both for preventing or correcting joint deformities and to stabilise joints which are no longer properly supported by muscles.
Psychological support is useful to establish a pathway of help and acceptance of the disease.
