Genic Therapy for CMT2A: researchers from the Dino Ferrari Center involved in the research thanks to the support of the Mitofusin 2 Project Association.
Milan, March 4, 2019
Dr. Federica Rizzo, Dr. Monica Nizzardo, Prof. Stefania Corti, Prof. Giacomo P. Comi, Prof Nereo Bresolin
Dino Ferrari Center, University of Milan, I.R.C.C.S. Foundation Ca ’Granda Ospedale Maggiore Policlinico.
Our research project aims to develop a possible therapeutic approach for CMT2A, a sensorimotor polyneuropathy, characterized by the death of motor and sensory neurons. The pathology is caused by mutations in the Mitofusin 2 (MFN2) gene, that codes for the MFN2 protein, located in the membrane of a cell organelle, the mitochondria, which represents the power station of cells and performs essential biological functions. As yet, unfortunately, no resolutive therapy is available and very few research groups are involved in studying this disease. Gene therapy represents a promising strategy, since it is designed to correct the genetic cause underlying the disease itself. This type of strategy is giving excellent results in clinical trials with adeno-associated type 9 vectors (AAV9) for the common form of Spinal Muscular Atrophy (SMA), associated with mutations in the SMN gene (www.clinicaltrials.gov). The clinical trial is currently underway also at the Policlinico of Milan and is followed by our researchers. Therefore, taking advantage of the skills we have acquired in this sector, we propose to use the same type of approach also for the CMT2A. Nevertheless, the causes of this pathology are both the lack of the “healthy” gene, and the presence of the “sick” MFN2 protein are the cause of the pathology. Thus, this aspect of the disease requires, in anticipation of a therapy for the patients, to introduce the “healthy” gene -as it can be done for SMA or for other diseases only caused by the lack of a protein- but also to turn off the sick gene. Furthermore, the MFN2 protein, due to its important role in mitochondria function, must be present “in appropriate quantity”, neither too low nor too high (another aspect that for other proteins isn’t so essential, in relation to their function).
For all these reasons, the development of a gene therapy for CMT2A requires additional efforts in terms of number of experiments and time.
On the bases of these premises, our therapeutic strategy for CMT2A requires to:
administer two therapeutic vectors: the first one enables to turn off the “sick” gene, the second one allows to express the “healthy” gene. A possible alternative may be the production of a single, larger vector, able to perform both functions.
obtain optimal MFN2 levels (neither too low nor too high) in order to avoid the appearance of adverse effects.
We have already generated created the necessary constructs, as well as demonstrated the silencing of the “sick” MFN2 gene and the correct expression of the “healthy” MFN2 protein, through the use of the vectors described in point 1, both in cellular and in animal models of this disease. In particular, in the cellular model we observed a marked improvement of the pathological phenotype (Figure 1).
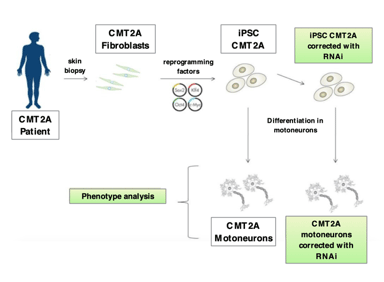
Figure 1. Evaluation of the impact of the therapeutic strategy on the cellular disease model.
We propose to complete these experiments and publish them in an indexed scientific journal, contributing to laying the first bases for an effective gene therapy for patients. Before proposing a clinical trial to the FDA/EMA, however, a more detailed analysis of this strategy with further studies in pre-clinical models is required. In this regard, the development of pre-clinical in vivo models of disease -that can more faithfully reproduce the clinical and neuropathological aspects of human CMT2A- will certainly be useful to assess the effectiveness of gene therapy.
The progress that has been made so far in this project will be presented in a session at the 71st Congress of the American Academy of Neurology, the largest professional organization of neurologists in North America, to be held on May 4-10, 2019 in Philadelphia. The presentation, entitled “RNAi/gene therapy combined approach and therapeutic strategy for Charcot-Marie-Tooth 2A” (Rizzo F, Bono S, Salani S, Bordoni A, Melzi V, Ruepp M, Pagliarani S, Barbullushi K, Abati E, Cordiglieri C, Bresolin N, Comi G, Nizzardo M, Corti S), will serve help to share the data and to focus the attention of a wide audience of researchers and clinicians on this disease. The commitment of our research group in recalling the scientific interest in this disease is also demonstrated by the recent publication in which we conducted a literature review concerning available in vitro and in vivo models of CMT2A and possible therapeutic approaches (Barbullushi K, Abati E, Federica Rizzo F, Bresolin N, Comi GP, Corti S. Disease modeling and therapeutic strategies in CMT2A: state of the art. in press Molecular Neurobiology).